Alternative TSS Usage
Philipp Ross
09-25-2018
Last updated: 2018-10-01
Code version: b1b8980
Using the cageR software package, we were able to detect around 70 genes with “shifting promoters.” We define this as a shift of at least 100 base pairs in a gene’s primary TSS between two points throughout the P. falciparum blood stage.
shifting <- readr::read_tsv("../output/ctss_clustering/modified/annotated_shifting.tsv")
shifting %>%
dplyr::mutate(shift=abs(groupX.pos-groupY.pos)) %$%
summary(shift) Min. 1st Qu. Median Mean 3rd Qu. Max.
100.0 158.2 206.5 231.0 272.2 772.0 shifting %>%
dplyr::mutate(shift=abs(groupX.pos-groupY.pos)) %>%
ggplot(aes(x=shift)) +
geom_histogram(color="grey70") +
ggtitle("TSS Shift")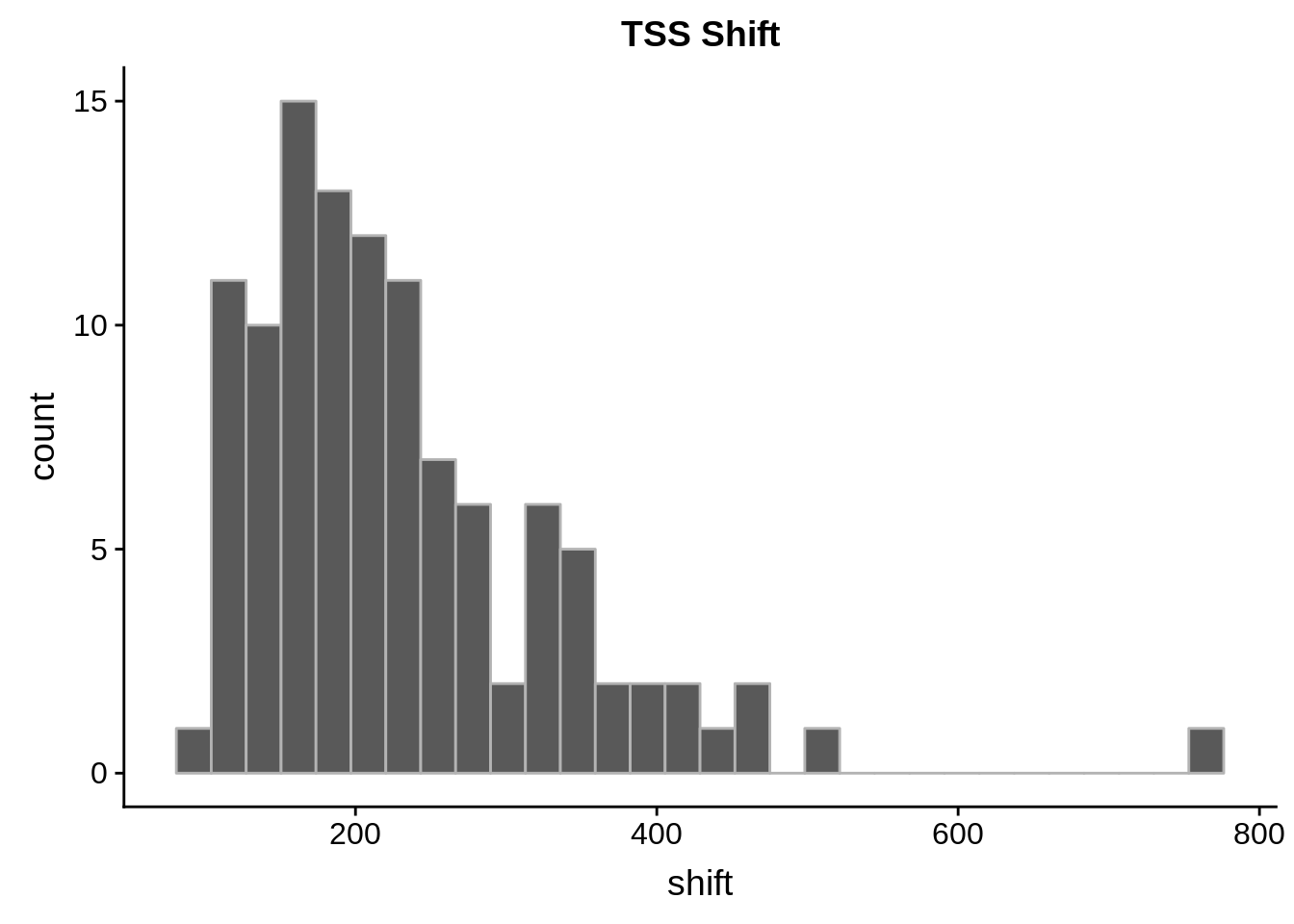
x3d7_abund <- readRDS("../output/neighboring_genes/gene_reduced_3d7_abund.rds")
pcg <- tibble::as_tibble(rtracklayer::import.gff3("../data/annotations/PF3D7_codinggenes_for_bedtools.gff"))$ID
get_filtered_ids <- function(abund,tpm_threshold) {
fabund <- abund %>%
dplyr::group_by(gene_id) %>%
dplyr::summarise(f=sum(TPM>=tpm_threshold)) %>%
dplyr::ungroup() %>%
dplyr::filter(f>0 & gene_id %in% pcg)
return(fabund$gene_id)
}
fx3d7 <- get_filtered_ids(x3d7_abund,5)
sx3d7_abund <- x3d7_abund %>%
dplyr::filter(gene_id %in% fx3d7) %>%
dplyr::select(gene_id,tp,TPM) %>%
dplyr::group_by(gene_id) %>%
dplyr::summarise(m=mean(TPM))
sx3d7_abund %>%
dplyr::filter(gene_id %in% shifting_genes) %>%
ggplot(aes(x=m)) +
geom_histogram(color="grey70") +
ggtitle("Mean Gene Expression")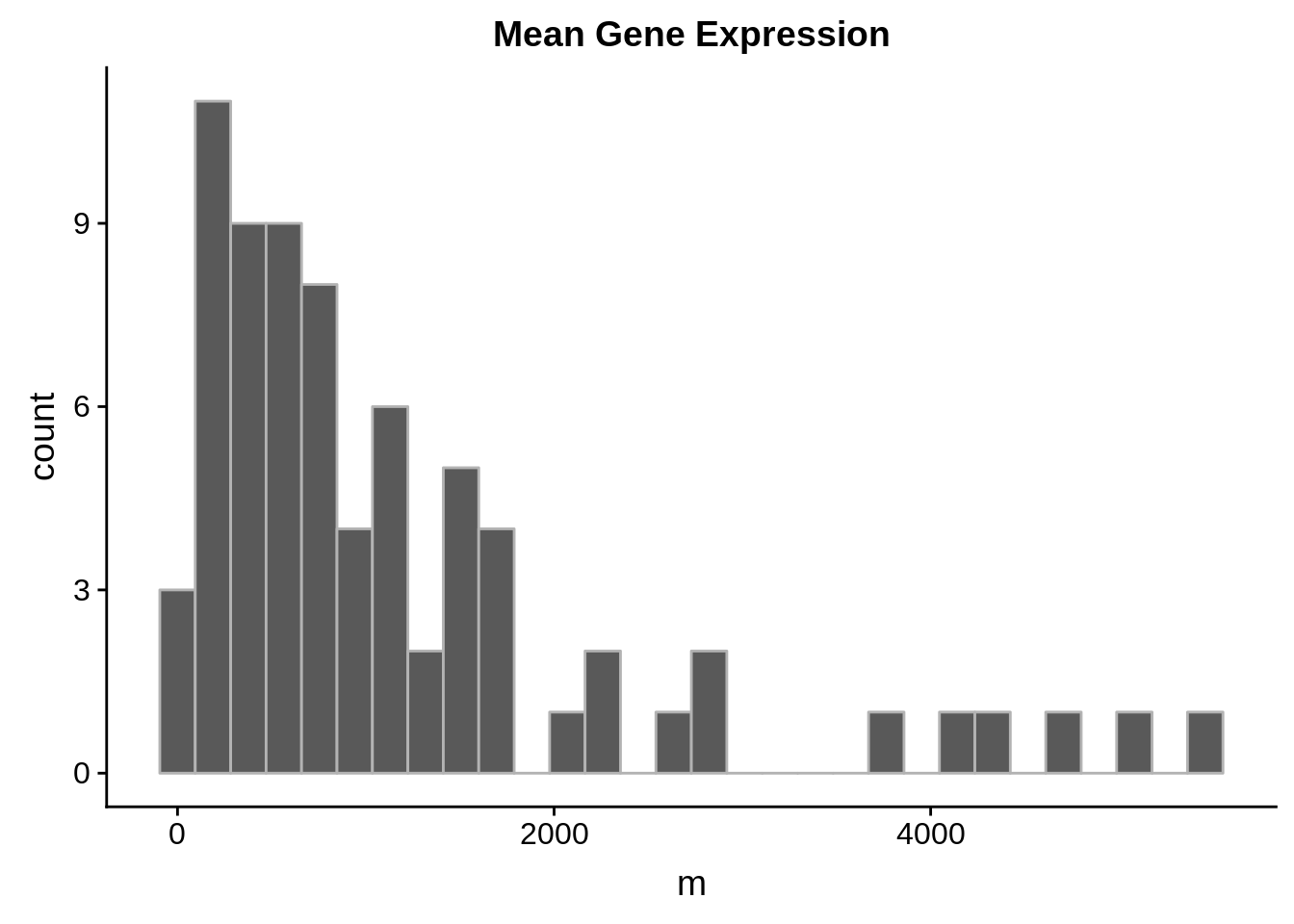
Session Information
R version 3.5.0 (2018-04-23)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Gentoo/Linux
Matrix products: default
BLAS: /usr/local/lib64/R/lib/libRblas.so
LAPACK: /usr/local/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] bindrcpp_0.2.2
[2] BSgenome.Pfalciparum.PlasmoDB.v24_1.0
[3] BSgenome_1.48.0
[4] rtracklayer_1.40.6
[5] Biostrings_2.48.0
[6] XVector_0.20.0
[7] GenomicRanges_1.32.6
[8] GenomeInfoDb_1.16.0
[9] org.Pf.plasmo.db_3.6.0
[10] AnnotationDbi_1.42.1
[11] IRanges_2.14.10
[12] S4Vectors_0.18.3
[13] Biobase_2.40.0
[14] BiocGenerics_0.26.0
[15] scales_1.0.0
[16] cowplot_0.9.3
[17] magrittr_1.5
[18] forcats_0.3.0
[19] stringr_1.3.1
[20] dplyr_0.7.6
[21] purrr_0.2.5
[22] readr_1.1.1
[23] tidyr_0.8.1
[24] tibble_1.4.2
[25] ggplot2_3.0.0
[26] tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] nlme_3.1-137 bitops_1.0-6
[3] matrixStats_0.54.0 lubridate_1.7.4
[5] bit64_0.9-7 httr_1.3.1
[7] rprojroot_1.3-2 tools_3.5.0
[9] backports_1.1.2 R6_2.2.2
[11] DBI_1.0.0 lazyeval_0.2.1
[13] colorspace_1.3-2 withr_2.1.2
[15] tidyselect_0.2.4 bit_1.1-14
[17] compiler_3.5.0 git2r_0.23.0
[19] cli_1.0.0 rvest_0.3.2
[21] xml2_1.2.0 DelayedArray_0.6.5
[23] labeling_0.3 digest_0.6.15
[25] Rsamtools_1.32.3 rmarkdown_1.10
[27] R.utils_2.6.0 pkgconfig_2.0.2
[29] htmltools_0.3.6 rlang_0.2.2
[31] readxl_1.1.0 rstudioapi_0.7
[33] RSQLite_2.1.1 bindr_0.1.1
[35] jsonlite_1.5 BiocParallel_1.14.2
[37] R.oo_1.22.0 RCurl_1.95-4.11
[39] GenomeInfoDbData_1.1.0 Matrix_1.2-14
[41] Rcpp_0.12.18 munsell_0.5.0
[43] R.methodsS3_1.7.1 stringi_1.2.4
[45] yaml_2.2.0 SummarizedExperiment_1.10.1
[47] zlibbioc_1.26.0 plyr_1.8.4
[49] grid_3.5.0 blob_1.1.1
[51] crayon_1.3.4 lattice_0.20-35
[53] haven_1.1.2 hms_0.4.2
[55] knitr_1.20 pillar_1.3.0
[57] XML_3.98-1.16 glue_1.3.0
[59] evaluate_0.11 modelr_0.1.2
[61] cellranger_1.1.0 gtable_0.2.0
[63] assertthat_0.2.0 broom_0.5.0
[65] GenomicAlignments_1.16.0 memoise_1.1.0
[67] workflowr_1.1.1 This R Markdown site was created with workflowr